La phylogénie, quelques concepts et définitions
Dans la cellule, l'ADN est transcrit en ARN messager dans le noyau chez les eucaryotes grâce à la RNA-polymérase. A ce stade l'ARN messager contient l'équivalent des parties codantes du gène, appelées exons, et des parties non-codantes, appelées introns. L'ARN messager subit alors plusieurs étapes de maturations, parmi lesquels l'épissage (qui excise les introns) et l'ajout d'une queue poly-A. L'ARN mature quitte le noyau pour être traduit en protéine dans le cytoplasme par les ribosomes (Philippe Gouret, r2)
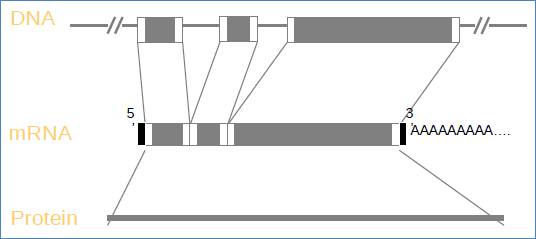
Figure 1: Gène et protéine (Philippe Gouret, r2)
L'annotation structurale consiste à détecter la position d'un gène sur une séquence d'ADN et plus précisément de la structure exons / introns qui le détermine et qui permet de prédire la ou les protéines pour lesquelles il code. Elle utilise des méthodes dites « ab initio », basées sur des critères statistiques concernant les propriétés différentes des régions codantes par rapport aux régions non-codantes des segments, ou des méthodes basées sur l'homologie, qui donc utilise la théorie de l'héritage avec modifications du matériel génétique des différentes espèces à travers l'évolution à partir d'ancêtres communs. Cette théorie a pour conséquence la conservation partielle des génomes des espèces à travers l'évolution, c'est-à-dire que les parties codantes sont mieux conservées que les parties non-codantes, puisqu'elles sont soumises à de plus fortes pressions de sélection (Philippe Gouret, r2)
À la différence de l'annotation structurale, l'annotation fonctionnelle consiste à prédire le plus précisément possible la ou les fonctions des protéines issues des gènes sur les plans: biochimique, cellulaire, métabolique et organique, avec plusieurs approches, dont la recherche de similitude statistique de séquences au niveau des acides aminés, et la phylogénie. Lorsque la similitude statistique de séquences est vérifiée de façon significative avec une autre séquence dont la fonction est présumée connue, elle semble impliquer une « fonction » similaire. La phylogénie s'appuie également sur une similitude de séquence, mais tente de modéliser mathématiquement l'histoire évolutive et donc les relations d'héritage entre les espèces et entre les gènes et protéines. Elle consiste alors à construire des arbres phylogénétiques retraçant l'histoire évolutive de séquences biologiques (protéines (séquences d'acides aminés), séquences nucléotidiques). Les noeuds de ces arbres regroupent, soit des séquences protéiques issues de gènes, soit des sous-arbres. Aux noeuds des sous-arbres sont associées des valeurs numériques indiquant leur solidité statistique (Philippe Gouret, r2)
La phylogénie introduit deux concepts permettant de différencier deux gènes proches, dits homologues: (Philippe Gouret, r2)
1. Les gènes homologues dérivant d'un gène ancestral par spéciation sont appelés orthologues et présentent des fortes probabilités pour posséder des comportements fonctionnels similaires.
2. Les gènes homologues produits par une duplication à l'intérieur du génome d'une même espèce sont appelés paralogues et s'orientent vers une divergence fonctionnelle, puisque la copie à travers le processus de l'évolution semble avoir adopté une nouvelle fonction.
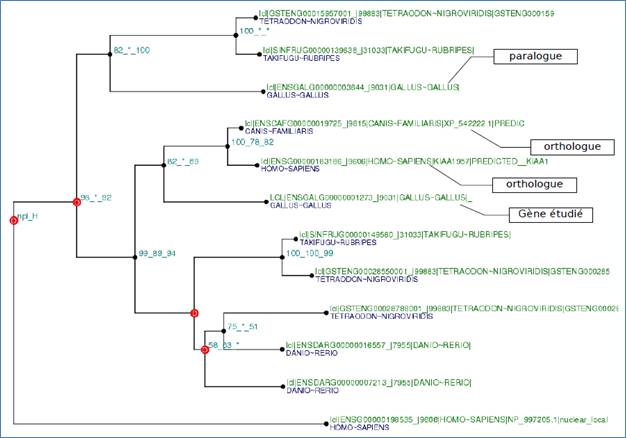
Figure 2: Orthologues et paralogues sur un arbre phylogénétique d'un gène étudié (Philippe Gouret, r2)
La phylogénomique applique des principes de l'analyse phylogénétique pour hiérarchiser l'évolution des espèces, mais propose l'utilisation de la totalité de l'information génomique, c'est-à-dire la structure du génome (Étienne Lord, r3)