Le problème des données de la bioinformatique
L'explosion de la génération de données génomiques et l'hétérogénéité de ces données de par les multiples logiciels de bioinformatique entraînent inévitablement un accroissement de l'écart entre les données, les connaissances et l'information que l'on peut en extraire.
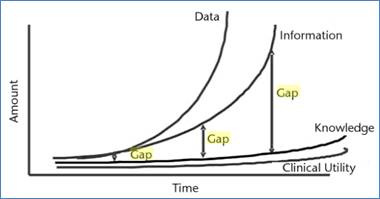
Figure 9: Relationship of data to information, knowledge, clinical utility, and the increasing gap between technology and science (Hai Hu, r14)
Les données de séquences du génome, de séquences de protéines, et de structure de protéines, en elles-mêmes sont d'une forte redondance, car un même fragment de séquence peut être présent dans plusieurs entrées, et leurs annotations sont peu normalisées, peu précises, sans compter des erreurs dans les annotations.
L'intégration des bases de données est difficile, puisque chaque base de données possède son propre format. Certains formats standards existent : asn.1, fasta, etc. mais aucun n'est universel. La différence de terminologies entre les différentes bases de données rend les requêtes vers de multiples bases très complexes.
Les outils informatiques développés et utilisés par les biologistes sont de natures très diverses, il peut s'agir des infrastructures informatiques, des programmes de gestion de linformation, ou encore des programmes informatiques de calculs, de corrélations, etc. À chaque outil est associé un nombre de paramètres, comme par exemple les méthodes d'inférence phylogénétique décrites auparavant. Il est souvent nécessaire pour les biologistes d'enchaîner ces outils manuellement, ce qui devient fastidieux lorsqu'il s'agit de le répéter pour modifier seulement quelques paramètres sur une large plage de données.
Les systèmes de gestion de flux de travaux sont alors utilisés afin de faciliter l'interaction entre ces outils, de rendre possiblement leur enchaînement, tout en visualisant de manière graphique l'architecture, le flux de données et l'exécution (Franck Valentin , r1)